Распространенность не зависит от гендерных различий, но варьируется в разных группах населения. Однако мальчики в возрасте до 1 года подвержены более высокому риску смерти.
Фенилкетонурия
Первые симптомы — частая рвота, рвота, экзема, судороги, плесневелый запах мочи и кожи. Ребенок может быть вялым или гиперактивным. Психомоторное развитие задерживается, могут наблюдаться признаки олигофрении. Диагноз может быть поставлен в родильном отделении. Все дети с фенилкетонурией определенно считаются «детьми с особыми потребностями».
Лечение состоит из специальной низкобелковой диеты, не содержащей продуктов FA.
Определение заболевания
Фенилкетонурия — это врожденное, генетическое нарушение гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и ее метаболитов, что приводит к тяжелым повреждениям центральной нервной системы. Впервые заболевание было описано норвежским врачом Й.А. Феллингом в 1934 году.
Когда болезнь была изучена, специалисты обнаружили, что за ее возникновение отвечает один-единственный ген фенилаланингидроксилазы. Первый успешный метод лечения был разработан и проведен в Англии в 1950 году.

В неонатальном периоде клиника отсутствует. Патология проявляется в первые шесть месяцев жизни ребенка. Затем накопление вещества приводит к тяжелым нарушениям развития. Поэтому чрезвычайно важно распознать дефект сразу после рождения и избегать употребления продуктов, содержащих фенилаланин. Последующее соблюдение диеты не устраняет дефект, но предотвращает развитие новых дефектов.
Патология одинаково распространена у обоих полов. Расовых отклонений не обнаружено. Большое количество пациентов наблюдается в таких странах, как Китай, Турция и Ирландия. В среднем по России. с фенилкетонурией рождается один из 7000 детей.
Причины фенилкетонурии
Существует три типа врожденных пороков развития, первый из которых считается классическим, так как диагностируется более чем в 90% случаев. Вторая и третья формы патологии встречаются реже. Симптомы схожи у всех типов, и заболевание приводит к умственной отсталости. В классической форме. фенилкетонурии можно избежать с помощью диетического лечения, в то время как атипичные варианты, к сожалению, не поддаются коррекции.
- Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
- Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
- Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивному типу. Это означает, что генетический дефект может быть унаследован от одного из родителей. Пол родителей и ребенка не играет роли.
Основной фактор риска фенилкетонурии — заключается в том, что у обоих родителей имеется дефект в гене PAH (фенилаланингидроксилаза). Заболевание развивается, когда оба родителя передают ребенку копию дефектного гена.
Классификация
Различают типичные (классические) и атипичные формы (вариантные, кофермент-зависимые, злокачественные — на них приходится около 10% случаев). фенилкетонурии в связи с генетической и клинической гетерогенностью нарушений метаболизма фенилаланина и его кофактора биотерола.
Причина второй атипичной формы диеторезистентности фенилкетонурии это дефицит дигидроптеридинредуктазы, который обусловлен дефектом гена на хромосоме 4p в положении 15.3 и был открыт Смитом в 1974 году. Дефект фермента нарушает восстановление активной формы тетрагидробиоптерина, кофактора гидроксилирования фенилаланина до тирозина и нейромедиаторов серотонина и катехоламинов, таких как L-DOPA и 5-окситриптофан.
Диетоустойчивая фенилкетонурия 3 была описана Кауфманом в 1978 году (согласно источнику Википедии) и характеризуется дефицитом синтетазы 6-пируват-тетрагидротерин, которая участвует в синтезе тетрагидробиоптерина.
Атипичные формы характеризуются прогрессирующими проявлениями заболевания.
Другие формы патологии связаны с аномалиями в других альтернативных путях метаболизма фенилаланина, что приводит к метилмандалевой ацидурии и парагидроксифеноксиуксусной ацидурии.
Причины возникновения фенилкетонурии
С увеличением опыта в диагностике и лечении фенилкетонурии Со временем стало ясно, что причиной этого наследственного заболевания являются мутации в гене фенилаланин-4-гидроксилазы — 12q23.2. Способ наследования — аутосомно-рецессивный.
Чаще всего заболевание вызывается резким снижением или отсутствием активности такого фермента печени под названием фенилаланин-4-гидроксилаза, который обычно необходим для катализа преобразования (гидроксилирования) фенилаланина в аминокислоту тирозин.
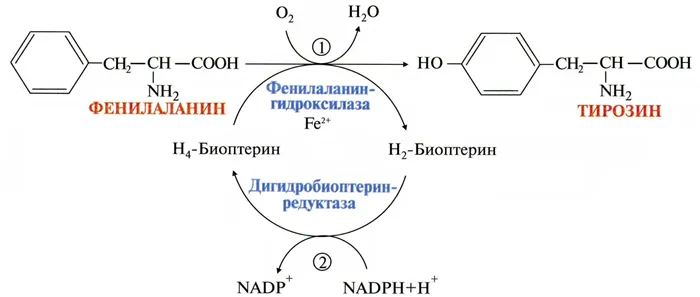
Метаболизм фенилаланина и тирозина
Однако около 10% случаев гиперфенилаланинемии обусловлены атипичными факторами, связанными с мутациями в других генах, кодирующих ферменты, участвующие в синтезе кофактора фенилаланингидроксилазы, известного как тетрагидробиоптерин (BH4).
Симптомы фенилкетонурии
Фенилкетонурия характеризуется довольно интенсивной клинической картиной, включающей такие симптомы, как:
- отставание физического развития с 6 месячного возраста;
- микроцефалия ;
- вегетативные дисфункции;
- повышенная возбудимость и двигательная гиперактивность; или дерматит, возможно с папулезными кожными высыпаниями (как на фото ребенка с фенилукетонурией);
- мышечная гипертензия;
- атаксия ;
- гиперкинезы ;
- неустойчивость походки;
- судорожные припадки;
- нередко обнаруживаются пороки сердца ;
- чувствительность к травматизации и осветление кожи, волос и радужки глаз ( депигментация ), в особенности при несоблюдении необходимой диеты, вызывающей недостаточность меланина в организме, являющегося производным тирозина.
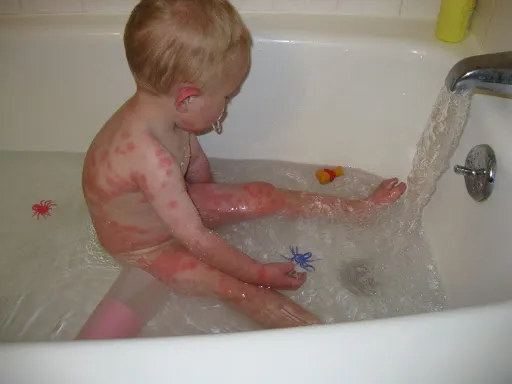
Экзема при фенилкетонурии
Психические нарушения
Фенилкетонурия вызывает значительные патологические изменения в обменных процессах в головном мозге, что приводит к следующим нарушениям:
- глубокая степень умственной отсталости, вплоть до идиотии или имбецильности ;
- трудности в обучении;
- возникновение явлений эхопраксии — повторение движений за окружающими;
- эхолалии (повторение речи);
- вялое поведение может сменяться редкими вспышками злости и раздражительности .
Как видно на фотографиях пациентов фенилкетонурией Их телосложение обычно диспластично, череп уменьшен в размерах, наблюдаются гипогонадизм и карликовость.
Основной фактор риска фенилкетонурии — заключается в том, что у обоих родителей имеется дефект в гене PAH (фенилаланингидроксилаза). Заболевание развивается, когда оба родителя передают ребенку копию дефектного гена.
Дефицит ферментов и патология

Фермент фенилаланин гидроксилаза вырабатывается клетками печени и некоторыми клетками в почках ребенка. Если организм не может вырабатывать достаточное количество этого фермента из-за дефекта гена, фенилаланин из пищи накапливается в организме и вызывает осложнения в органах и тканях. Поэтому если организм не может вырабатывать ферменты, он не может расщеплять фенилаланин из пищи, что в конечном итоге приводит к болезни. — фенилкетонурии.
Что вызывает фенилкетонурию: основа болезни
Ребенок может заболеть фенилкетонурией, если они наследуют дефектный ген от своих родителей. Дефектный ген приводит к неспособности вырабатывать фермент фенилаланин гидроксилазу. Заболевание наследуется по аутосомно-рецессивному типу. Другими словами: Если один из родителей наследует здоровый ген, а другой — дефектный, то здоровый ген доминирует над нездоровым рецессивным геном. При таком У ребенка не будет заболевания, но он будет нести копию дефектного гена в своей ДНК.
Если дефектный ген есть у обоих родителей, у ребенка есть 25-процентный шанс иметь FCU, 25-процентный шанс быть генетически здоровым и 50-процентный шанс унаследовать копию гена, как у родителей. Однако у них не наблюдается никаких симптомов заболевания. Ребенок может страдать от фенилкетонурии, если оба родителя имеют дефектные гены, которые они передают ребенку, или сами страдают от заболевания.
Фенилкетонурия является редким заболеванием и встречается у 1 из 10 000-15 000 новорожденных. Новорожденных тестируют сразу после рождения, чтобы лечение можно было начать как можно раньше.
Каковы симптомы фенилкетонурии у ребенка?
У ребенка, страдающего этим заболеванием, могут наблюдаться следующие симптомы:
- Заторможенность, вялость и необъяснимая общая слабость.
- Хронические срыгивания или приступы рвоты. Это может привести к обезвоживанию в качестве осложнения.
- Высыпания, которые постепенно распространяются на все большие участки кожи, что приводит к экземе.
- Раздражительность и суетливость. У подросших малышей с ФКУ возникают частые эпизоды вспышек гнева и могут проявляться признаки нарушений нервно-психического развития.
- Бледная кожа, светлые глаза и волосы из-за недостаточного количества меланина в организме.
- Затхлый запах от кожи, напоминающий мышиный, и аналогичный аромат мочи ребенка или пота из-за присутствия фенилуксусной кислоты.
- Могут возникать тремор и эпилепсия. Около 25% детей старшего возраста с недиагностированными проявлениями ФКУ имеют олигофрению.
Снижение роста и веса ребенка, нарушение обменных процессов, задержка физического и психического развития. Симптомы не проявляются, пока ребенок еще очень маленький, первые симптомы появляются в возрасте трех месяцев. В некоторых случаях симптомы не проявляются до шестимесячного возраста, когда ребенок начинает есть твердую пищу. Визит к врачу может помочь родителям узнать больше о симптомах.